Molecular Docking
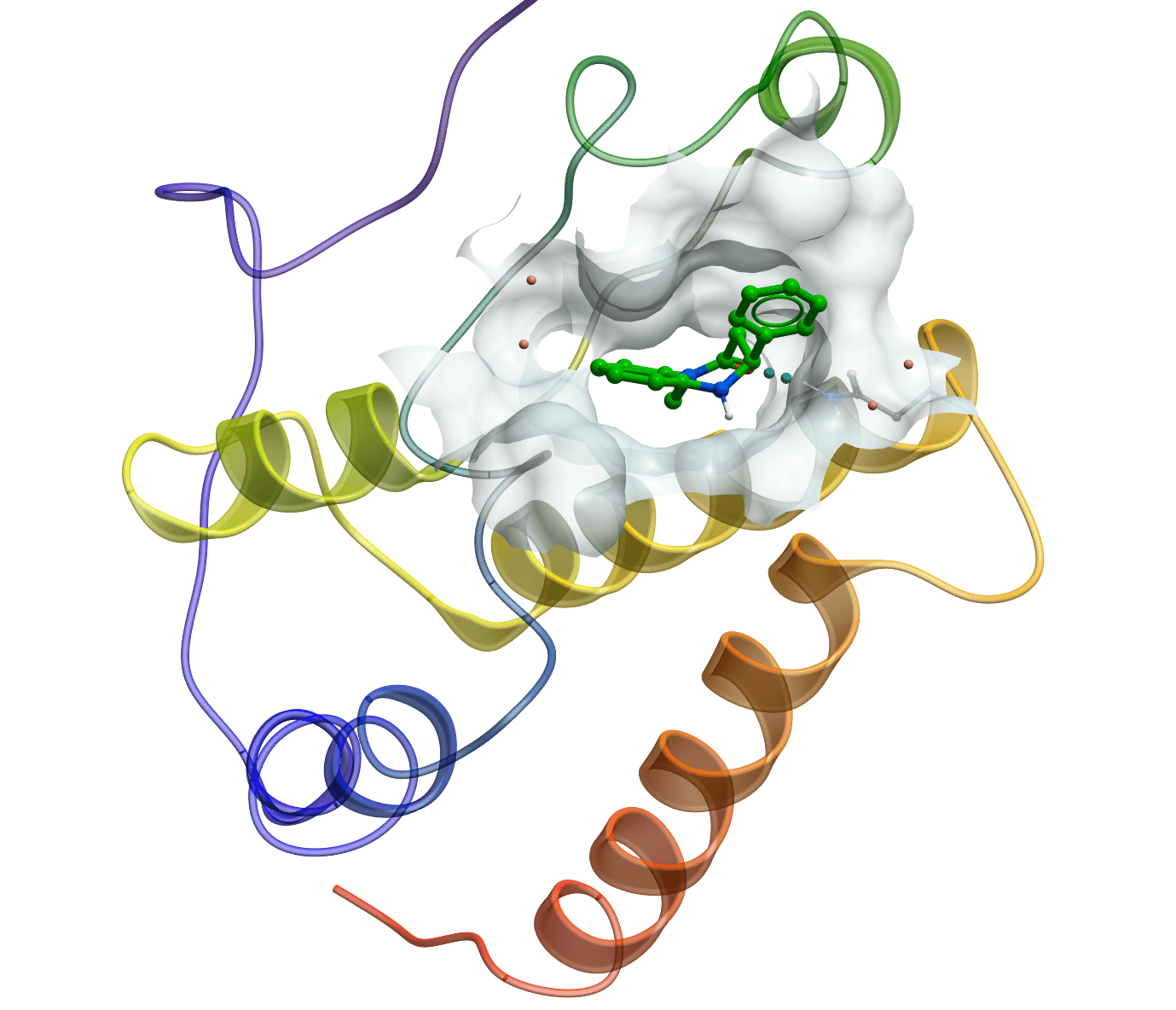
Protein-ligand interactions predicted by molecular docking
Molecular docking plays an essential role in structure-based drug discovery, with the aim to understand protein-ligand interaction precisely at the atomic level. LeDock is designed for fast and accurate flexible docking of small molecules into a protein. It achieves a pose-prediction accuracy of greater than 90% on the Astex diversity set and takes about 3 seconds per run for a drug-like molecule. It has led to the discovery of novel kinase inhibitors and bromodomain antagonists from high-throughput virtual screening campaigns. It directly uses SYBYL Mol2 format as input for small molecules. The graphic version on Windows operating system greatly simplifies common multiple sophisticated docking procedures for medicinal chemists.
LeDock exhibits an outstanding performance in a recent comprehensive evaluation of docking programs on a diverse set of 2002 protein-ligand complexes (Phys. Chem. Chem. Phys. 2016, 18, 12964).
- High accuracy in pose prediction
- User friendly for both virtual screening and hit elaboration
- Very fast
Fragment-Based Drug Design
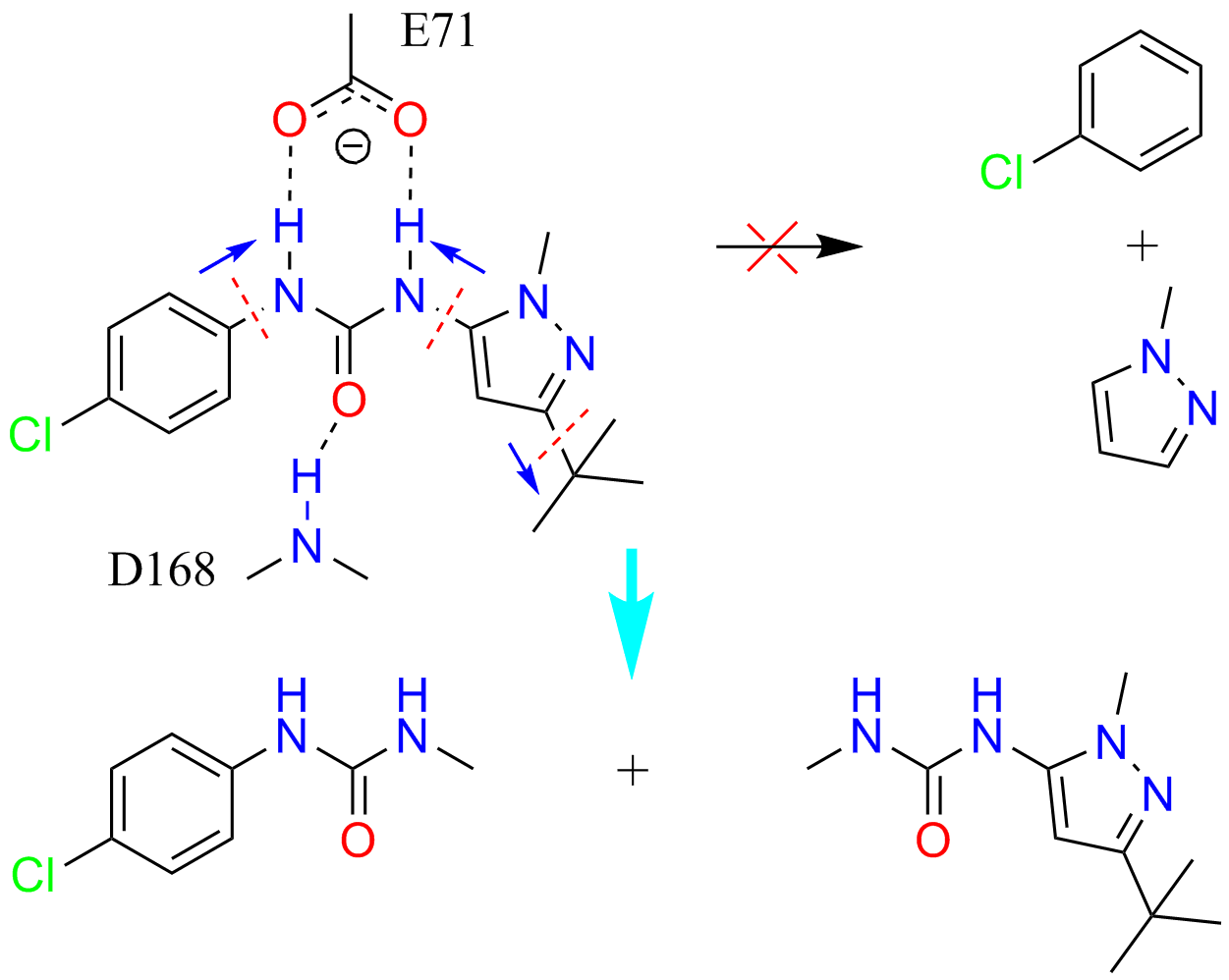
Schematic illustration of automatic fragmentation of a type II kinase inhibitor
Drug discovery is plagued by high attrition rates at all stages in research and development. Attrition, at least in part, can be traced back to the quality of core fragments. Fragment-based drug discovery (FBDD) is increasingly popular in both academia and pharmaceutical industry, for reducing attrition and offering leads even for previously intractable biological targets.
LeFrag is designed for in silico FBDD, with a pharmacophore oriented fragmentation algorithm. Its functions include automatic fragmentation of a compound library, similarity search, fragment-based core scaffold hopping, pharmacophore filtering, and substructure search.
- Fragments with high chemical richness
- Statistics on frenquency of fragments
- Unique core-fragment hopping
- Basic chemoinformatic analysis
- User friendly and extremely fast
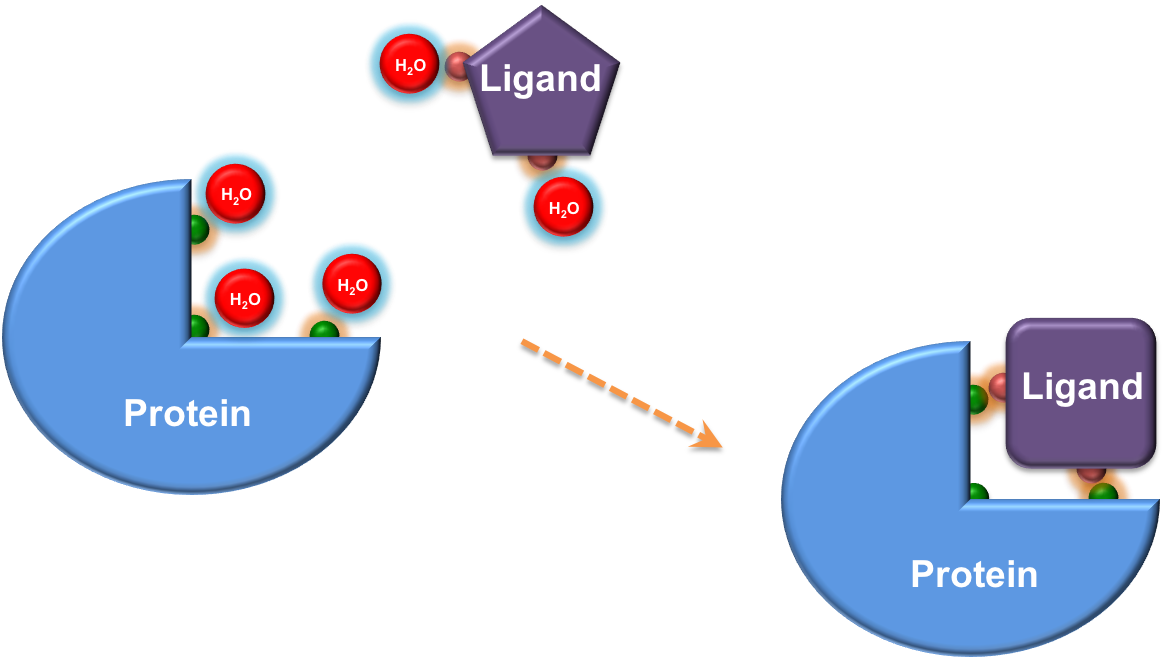
Accounting for Water
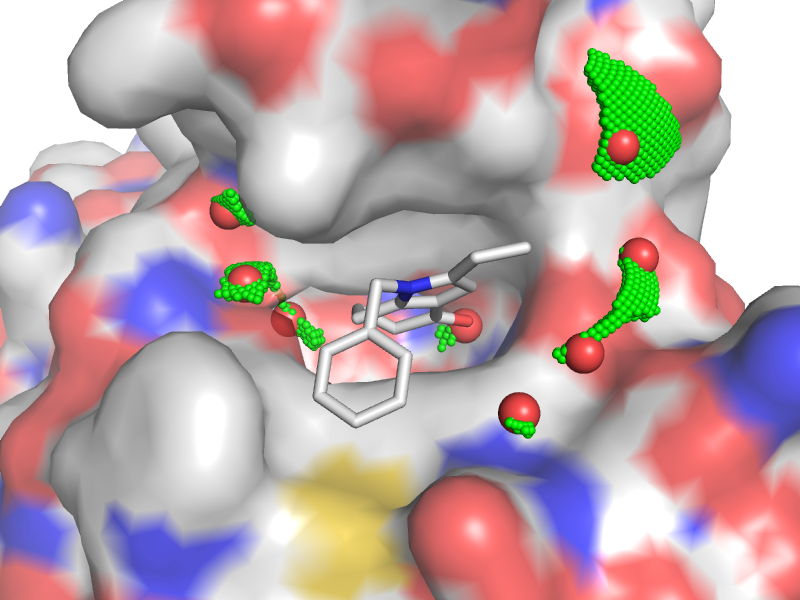
Acetyl-lysine recognition site of BRD4 bromodomain (PDB code 4PCE) with the crystal water molecules (red) and computed water clusters (green)
LeWater is designed to detect unfavorable polar interactions, constrained crystal water and hydrogen bonding penalty at the protein-ligand interface.
Hydrogen bonds are very important for binding specificity, but can be made by the ligand either in the protein or in solution. In order to form hydrogen bonds between the ligand and the protein, donors and acceptors firstly have to break hydrogen bonds with water, and then form new hydrogen bonds. In this sense, the enthalpic gain by formation of new hydrogen bonds is marginal and the gain is mainly entropic due to releasing waters into bulk. However, if newly formed hydrogen bonds between the ligand and the protein are not in good geometry or desolvated donors/acceptors are not involved into new hydrogen bonds, an enthalpic loss arises due to uncompensated breakage of hydrogen bonds with waters.
The program is developed to calculate the hydrogen bonding penalty index in the nature of enthalpy by checking both the solvation and hydrogen bonding state of donors/acceptors at the protein-ligand interface. Further integrating the hydrogen bonding penalty into a scoring function, it is found that a value of 1 in penalty corresponds to about 1.7 kcal/mol in binding free energy, which is in good agreement with the experimental value, e.g., breakage of a neutral hydrogen bond resulting in loss of energy from 0.5 to 1.5 kcal/mol.
The program can also be used to generate a water map at the protein-ligand interface to facilitate hit-to-lead optimization.
- Automatic dectection of hydrogen bonds at the protein-ligand interface
- Calculation of hydrogen bonding fitness and hydrogen bonding penalty
- Probing the constrained crystal water
- User friendly in virtual screening and structure-based drug design
Explicit Hydrogen in Proteins and/or Nucleic Acids
LePro is designed to automatically add hydrogen atoms to proteins and/or nucleic acids by explicitely considering the protonation state of histidine. All crystal water, ions, small ligands and cofactors except HEM were removed. It also generates a docking input file for LeDock with the binding site determined as to include any protein atom within 0.4 nm of any atom of the first complexed small molecule.
- Explicitely accounting for different protonation state of histidine
- Reducing modified residues
- Writing a docking input file for LeDock
- User friendly
Scoring Function
Key elements underlying the binding of a ligand to its receptor: shape complementarity, hydrogen bonding penalty, and ligand conformational strain
Scoring function, which predicts the binding affinity of a ligand to its receptor, is at the center of drug discovery, from hit identification to lead optimization. Fully accounting for water together with ligand conformational strain upon binding, LeScore demonstrates the superior performance in pose discrimination and accurate prediction of binding affinity.
Prospectively, LeScore has been extensively validated in high-throughput virtual screening campaigns against a variety of drug targets from kinases to epigenetic readers of bromodomains. Very recently, LeScore has been significantly improved in terms of both computational speed and general transferability. It shows a correlation coefficient of 0.84 on a subset of the PDBBind core data set.
QueryDB
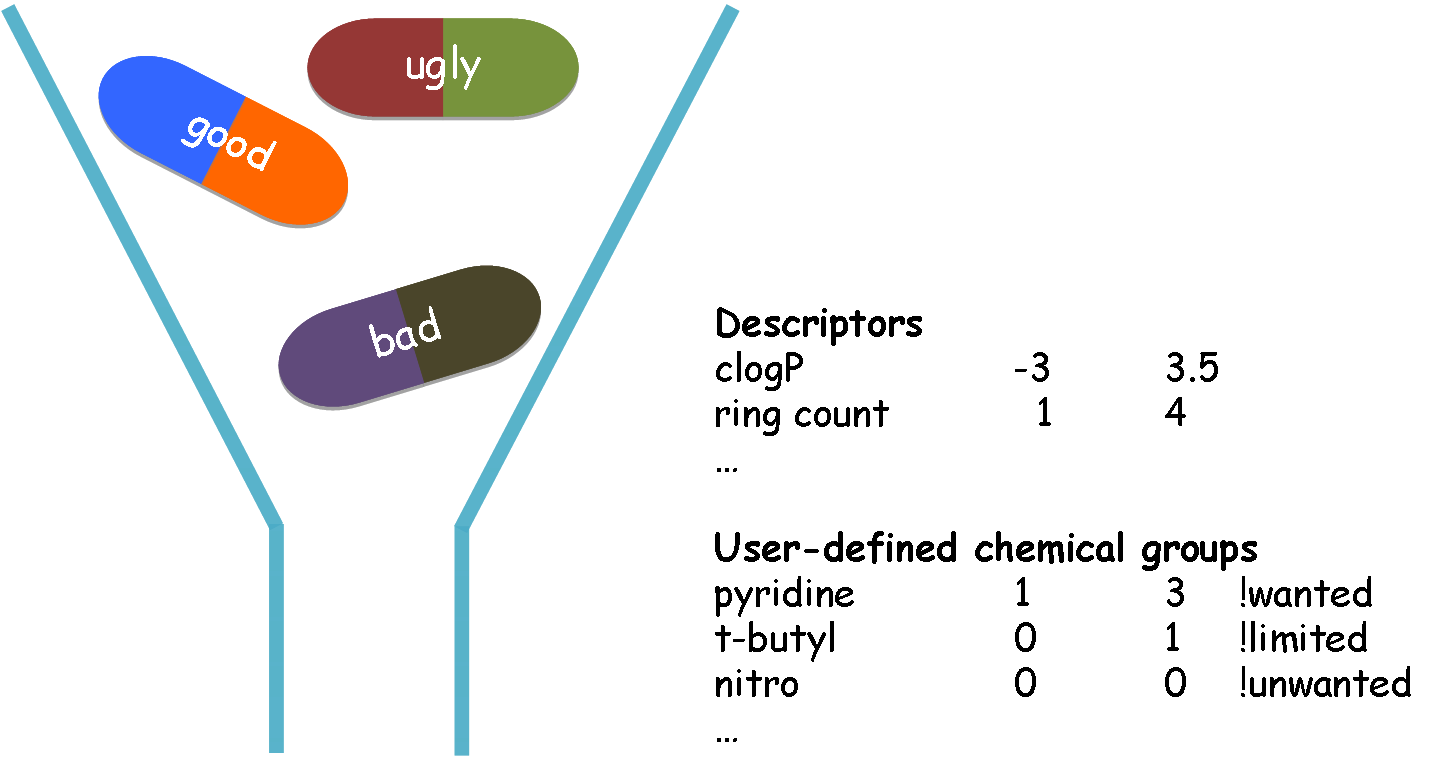
Schematic illustration of retrieving molecules from a library
QueryDB is a very fast and flexible molecular filtering and selection tool. It uses a combination of physical property calculations and functional group knowledge to: 1) remove undesirable compounds before they enter experimental or virtual screening, 2) retrieve molecules with preferred functional groups, and 3) substructure search.
Eliminating unwanted molecules before the use of modeling applications will substantially increase the computational efficiency, and the positive predictive value of such tools by remvoing false positives. Undesirable properties may include: toxic functionalities, a high likelihood of reacting with the target protein, and/or a low probability of oral bioavailability. Example of user-defined filtering criteria
ClusterByMCS
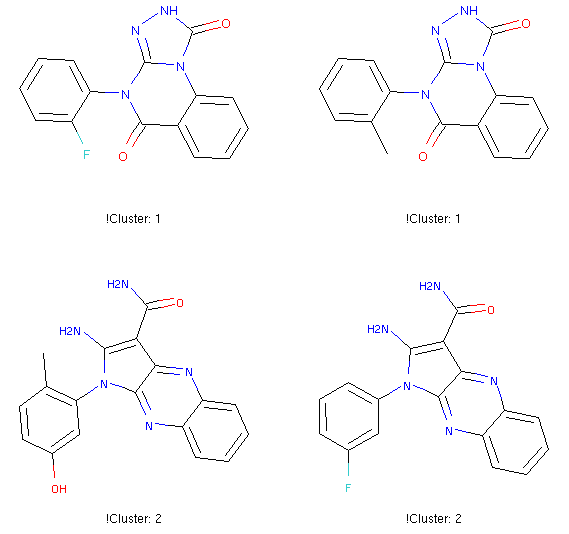
Clustering by maximum common substructure (MCS)
ClusterByMCS clusters molecules into groups according to maximum common substructure. Chemists can easily get an idea of chemotypes by eye-browsing very few moelcules. However, with hundreds of bioactive molecules from a HTS, it is non-trivial. This tool can automate what chemists have been doing by eyes so far, for easy analysis of HTS data to spot good scaffolds for future expansion, or analysis of structure-activity relationship.